|
définition et causes pathophysiologiques
Le prurit est défini cliniquement comme la sensation subjective et désagréable d’un désir de se gratter. Il y a une composante sensorielle débutant dans la peau, qui transite par les ganglions dorsaux et est traitée dans le système nerveux central. Souvent, la réaction motrice suit sous la forme d’un désir de se gratter. Le fait de se gratter abîme la peau et déclenche une réaction inflammatoire, que renforce encore la démangeaison. Le cercle vicieux typique commence. La peau est la frontière extrême du système nerveux et par conséquent richement innervée. La densité des fibres nerveuses cutanées varie selon les différentes parties du corps.1 De récentes études montrent que les terminaisons nerveuses libres de l’épiderme peuvent être classées dans deux catégories. D’une part, les terminaisons nerveuses «peptidergiques» contenant la substance P et la CGRP (calcitonin gene related peptide), qui débutent dans la couche granuleuse, d’autre part, les fibres nerveuses «non peptidergiques» qui n’ont pas de substance P ou CGRP, mais qui présentent, par contre, le marqueur Mrgprd (mass related G-protein).2 Ces fibres nerveuses commencent directement sous la couche cornée. Ces deux types de fibres nerveuses de l’épiderme se projettent dans différentes régions voisines de la moelle épinière. Les tâches précises de ces deux fibres nerveuses ne sont pas encore connues. Mais, si ces deux différentes fibres (nerveuses) transmettent des sensations de douleurs différentes, l’une d’elles pourrait probablement être responsable de la démangeaison. Sur la base de telles réflexions, nous avons récemment émis l’hypothèse des «couches».3 Ceci signifie qu’un faible stimulus suggère et déclenche une démangeaison des fibres nerveuses superficielles, alors qu’au contraire un fort stimulus excite les fibres nerveuses profondes de la zone dermo-épidermique, ceci étant ressenti comme douleur. Les fibres nerveuses évoquant un sentiment de douleur pourraient débuter dans la couche profonde de l’épiderme et dans ce cas le sentiment de démangeaison commencerait dans la couche superficielle de la peau. La perception de la douleur semble empêcher le sentiment de démangeaison au niveau des ganglions dorsaux et du SNC. Les patients avec de fortes démangeaisons s’arrachent pratiquement la peau en se grattant. La douleur ressentie est habituellement mieux tolérée que le prurit. La démangeaison est considérée comme un symptôme subjectif difficilement quantifiable et est physiologiquement difficile à saisir, surtout dans les expérimentations animales. Schmelz et coll.4 ont pu isoler les fibres C spécifiquement histaminiques, celles qui ne dépendent pas des fibres de la douleur. L’histamine n’est pas l’unique substance provoquant des démangeaisons. Le tableau 1 montre un choix des substances déclenchant le prurit. Cette liste incomplète comprend des neuropeptides, des neurotransmetteurs, des enzymes, différentes formes de cytokines et de médiateurs d’inflammation.5 Ceci démontre aussi combien les formes de démangeaisons sont multiples et que la thérapie ne peut pas se limiter à utiliser exclusivement des antihistaminiques.

Des expériences cliniques nous indiquent que lors de démangeaisons aiguës, une thérapie antihistaminique est mieux appropriée. Alors qu’au contraire, lors de démangeaisons aiguës et chroniques, l’histamine semble jouer un rôle défavorable. Lors de démangeaisons chroniques, une modification des récepteurs de neuropeptides, un changement de densité et de morphologie des fibres C apparaissent dans l’épiderme.3 Des modifications des fibres C sensorielles et périphériques ont également été observées lors de douleurs chroniques symptomatiques.6 Les modifications chroniques des terminaisons nerveuses périphériques conduisent dans la recherche de la douleur au phénomène d’allodynie. Un phénomène semblable existe avec la dermatite atopique, l’alloknesis. Alloknesis veut dire, que lors d’un stimulus mécanique ordinaire ou douloureux, un désir de se gratter est ressenti.7 Des sentiments de douleur induits par stimulations électriques transépidermiques ou par piqûres seront ressentis, chez les sujets atopiques, comme une démangeaison. Ceci est également valable pour une fine irritation mécanique des fibres de laine qui conduit également à un sentiment de démangeaison. Ce sentiment amène les individus atopiques à l’aversion typique de la laine. L’ancien nom «névrodermite», remplacé par «dermatite atopique» par analogie, n’est par conséquent pas déplacé puisque avec les atopiques, non seulement l’homéostasie et le système immunitaire de la peau sont modifiés, mais également le système nerveux cutané. En résumé, on doit retenir qu’avec des douleurs et des démangeaisons chroniques, le traitement se fera sur une longue période, afin de pouvoir rétablir les modifications au niveau du système nerveux cutané.
diagnostic différentiel
Il est utile de faire la distinction entre un prurit avec et un prurit sans exanthème. Il est par conséquent essentiel que le patient soit examiné de la tête aux pieds (y compris les muqueuses). En outre, il est également important de faire le lien temporel, d’évaluer l’intensité, les déclencheurs et la localisation du prurit, ainsi que l’anamnèse personnelle, familiale et médicamenteuse du patient. Toutes ces pièces de puzzle peuvent contribuer à donner une image complète et vraisemblablement mener à la problématique de base de ces démangeaisons. Dans les tableaux 2 et 3, on trouve une classification de la publication de Ward et Bernhard que j’ai adaptée 8 et qui devrait faciliter la réflexion sur les diagnostics différentiels. En cas de prurit sévère, plusieurs causes sont généralement combinées. Ainsi, une maladie de la glande thyroïde peut aggraver le prurit lié soit à un dessèchement de la peau des personnes âgées, soit à une diathèse atopique. En outre, des démangeaisons sans exanthème primaire, comme une xérose cutanée secondaire, peuvent mener à un eczéma de dessèchement avec des modifications inflammatoires de la peau.


prurit avec exanthème
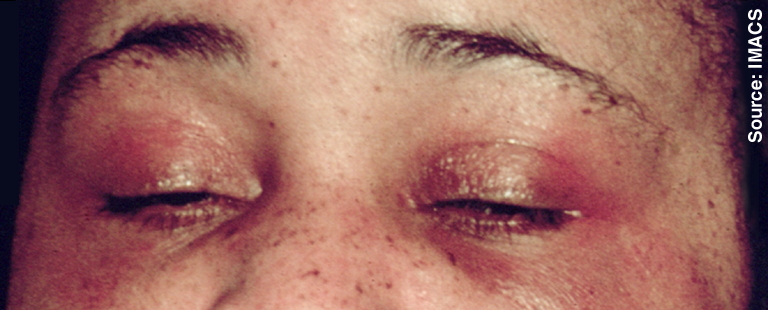
Les maladies eczémateuses de la peau représentent la cause la plus fréquente des démangeaisons avec exanthème. Les eczémas peuvent avoir différentes causes et apparaître sous forme locale ou généralisée. Cliniquement, on distingue fondamentalement l’eczéma aigu de l’eczéma chronique. Un eczéma aigu ou subaigu se traduit par des rougeurs, des papules, des vésicules et provoque de fortes démangeaisons. Lors d’un eczéma chronique, la peau est épaissie et lichénifiée, probablement en raison d’un grattage incessant et d’une inflammation chronique, ces démangeaisons répondent mal à une thérapie antihistaminique. Les démangeaisons chroniques portent généralement davantage atteinte à la qualité de vie du patient que les démangeaisons aiguës, celles-ci le dérangeant nuit et jour. Ce genre de démangeaisons, telles que le prurit chronique lors de dermatite atopique, nécessitent un défi thérapeutique particulier. Ici également, l’histamine semble jouer un rôle mineur.9 D’autres mécanismes semblent être impliqués dans la pathogenèse des démangeaisons (opioïdes endogènes, acétylcholine ou substance P). Il est toujours question de savoir si le lichen simplex chronique de Vidal représente une variante localisée de la névrodermite ou s’il s’agit d’une entité propre. Cliniquement, le lichen simplex est caractérisé par des démangeaisons chroniques, circonscrites et accompagnées d’un épaississement lichénoïde. Les endroits de prédilection sont souvent périanal (figure 1), périgénital, sur la nuque ou le bas des jambes. Ici également, le patient est dérangé nuit et jour en raison de démangeaisons chroniques et récurrentes.
Le prurit, lors d’urticaire chronique et aiguë aussi bien que lors de mastocytose, semble être déclenché principalement par des émissions d’histamine. Par conséquent et dans ce cas, un traitement antihistaminique est en règle générale efficace.
Le prurigo simplex subaigu apparaît comme un symptôme de différentes maladies. Le prurit est fréquemment associé au diabète (mellitus) ou à d’autres maladies endocriniennes. Cliniquement, on remarque des papules érosives et grattées sur tout le corps, dont les lésions initiales sont les séro-papules. Il est intéressant de constater que ces modifications de la peau ne se trouvent pas dans des endroits du corps inatteignables par le patient. Ici également, la composante traumatique et mécanique du grattage semble jouer un rôle principal. Le besoin impératif de se gratter conduit secondairement à une modification de la peau. Une pemphigoïde bulleuse devrait constamment être envisagée lors de formes chroniques de démangeaisons avec modifications érosives de la peau.

prurit sans exanthème
Un diagnostic différentiel de prurit sans exanthème représente un grand problème puisque aucune modification primaire de la peau n’apparaît et qu’ainsi aucune piste de diagnostic n’est donnée. La cause la plus fréquente et souvent sous-estimée pour ce genre de prurit est le prurit sénile. Cliniquement, on trouve une sécheresse cutanée, à peine ou pas de rougeur, une peau craquelée et lézardée, avec des endroits d’eczéma secondaire au grattage. La pathogenèse du prurit à cause d’une peau sèche n’est pas encore clarifiée. En raison des fissures de l’épiderme, les terminaisons nerveuses épidermiques superficielles sont directement exposées à l’air, ce qui pourrait directement en modifier la sensibilité et la conductivité. On peut réduire les sensations de démangeaisons en appliquant une crème grasse, celle-ci fonctionnant comme une occlusion étant donné que les fibres nerveuses ne sont plus exposées à l’air. L’expérience clinique quotidienne montre également que l’usage de crème grasse dans une thérapie de soins est l’une des meilleures thérapies contre les démangeaisons.
La défaillance rénale chronique et non pas aiguë déclenche une forte démangeaison résistante aux thérapies. Les démangeaisons sont indépendantes de l’âge, de la race, du sexe et de la pathologie rénale. Les mécanismes précis ne sont pas encore connus, mais il semble que les substances résiduelles de déchets ne pouvant plus être filtrées par les reins s’accumulent dans la peau. Environ 40% des patients avec une défaillance rénale chronique déclenchent un prurit urémique. Généralisé pour 19% d’entre eux, disséminé pour 50% et localisé sur une partie du corps pour 31%.10 Plus un patient requiert de dialyses, plus le risque de développer des démangeaisons chroniques existe. Il est intéressant de constater que la fréquence des démangeaisons est liée au type de membranes utilisées lors des dialyses. Des membranes de polysulphon déclenchent apparemment plus de démangeaisons que des membranes d’hémophane ou de cuprophane. Evidemment ces facteurs devront être étudiés plus encore. En outre, il est important de mentionner que le dessèchement de la peau joue un rôle important dans la formation du prurit rénal.
Le prurit hépatogène se déclenche avant tout lors d’une cholestase, les substances résiduelles de déchets semblent ici également se déposer dans la peau. Avec le prurit hépatogène, des opioïdes endogènes jouent un rôle important. Ainsi, lorsque l’on traite un prurit de cholestase par des antagonistes opioïdes systémiques, le patient peut déclencher un symptôme de sevrage. En outre, lors d’injections intradermiques de plasma de patients avec prurit hépatogène aux singes, ces derniers développent un prurit local naloxone réversible.11
Des hypo- et également des hyperthyréoses déclenchent des démangeaisons, parfois lors d’une xérodermie. Le diabète est également une cause endocrinienne importante dans le déclenchement de démangeaisons. Bien que personne ne nie que le diabète soit impliqué dans des formes de démangeaisons locales et généralisées, il n’y a pas d’études épidémiologiques convaincantes complètes et les options thérapeutiques correspondantes sont encore limitées.
Mis à part le syndrome de Sézary manifesté par une érythrodermie, d’autres lymphomes et maladies myélo-prolifératives peuvent également déclencher des démangeaisons. Ces démangeaisons ne montrent généralement pas de modification de la peau. Cinquante pour cent des patients avec une Polycythaemia vera rubra souffrent de démangeaisons massives, en particulier après une douche. Dix pour cent des patients avec un prurit aquagénique développent une Polycythaemia vera rubra. Lors de prurit aquagénique, le contact de l’eau provoque des démangeaisons. Ici, également, on ne peut faire que des spéculations, puisque les neurotransmetteurs, comme l’acétylcholine, semblent jouer un rôle.12
Différentes tumeurs malignes de type solide peuvent provoquer des démangeaisons dans le sens d’un syndrome paranéoplasique. Des indices montrent que des démangeaisons dans des endroits particuliers peuvent être déterminantes pour une tumeur solide cachée. Par exemple, un prurit scrotal peut dévoiler un carcinome de la prostate, des démangeaisons vulvaires un carcinome du col de l’utérus et des démangeaisons périanales un carcinome rectal.
Différents médicaments peuvent déclencher des démangeaisons massives avec ou sans exanthème (par exemple : opioïdes, paracétamol, antibiotiques, inhibiteurs de l’enzyme de conversion). Lors de la plupart des formes de démangeaisons déclenchées par des médicaments, un processus immunologique est en cause. Mais, quelques médicaments, tels que les opioïdes peuvent déclencher des crises de démangeaisons par une stimulation directe des fibres nerveuses. Des démangeaisons associées au HES (Hydroxyethyl-starch) nous indiquent que le HES s’accumule dans les terminaisons nerveuses périphériques.13
Finalement, les démangeaisons doivent être mentionnées lors de maladies psychiatriques. Les démangeaisons avec psychose, en particulier les illusions parasitaires, sont les plus compliquées à traiter. Une maladie névrotique peut également entretenir des démangeaisons ano-génitales. Le prurit sine materia, souvent évoqué, est certainement un diagnostic d’exclusion, dont la cause primaire n’a généralement pas été trouvée ou qu’une affection psychiatrique n’a pas été décelée. Des combinaisons peuvent certainement apparaître où une maladie psychiatrique peut renforcer les démangeaisons primaires ou inversement.
traitement
En principe, la thérapie contre le prurit est déterminée en fonction de la maladie de base. Par conséquent, il est très important d’appliquer une thérapie symptomatique, compatible avec un diagnostic précis. La mesure primaire la plus importante contre le prurit est une thérapie de soins hydratants en crème grasse ayant une base aussi neutre que possible. Je voudrais parler très clairement d’une thérapie de soins, que mes propres expériences ont démontré avec une étude en double aveugle et placebo contrôlée, qu’une base hydrante grasse réduit la sensation de démangeaisons en une à deux heures. Cette base devrait contenir aussi peu que possible d’additifs (par exemple : matières odorantes, agents conservateurs), qui pourraient irriter ou provoquer des allergies de la peau et ainsi renforcer les démangeaisons. La thérapie de soins hydratants en crème grasse devrait être appliquée plusieurs fois par jour, adaptée à la sécheresse clinique de la peau. De plus, les produits nettoyants de la peau non gras ou agressifs, ainsi que les produits à base d’alcool ou de gel sont à éviter.
Les antihistaminiques jouent encore le plus grand rôle dans les maladies dermatologiques en relation avec le prurit. On doit toutefois souligner que dans le cadre d’une dermatite chronique atopique, d’un lichen simplex chronique et d’un prurigo simplex sous-cutané, mais également dans le cadre d’un lymphome cutané, cette thérapie est insuffisante, elle est souvent utilisée faute d’alternative. Il n’y a pas d’études convaincantes, qui prouveraient que les antihistaminiques agissent effectivement lors de ces formes de prurit. Seuls d’anciens antihistaminiques, avec un effet sédatif, ont souvent eu une certaine efficacité. L’effet antiprurigineux d’antihistaminiques récents, non sédatifs est incertain et doit encore être soumis à de grandes études. Des stéroïdes internes ont un effet anti-inflammatoire et réduisent également les démangeaisons à court terme. En général, le prurit récidive lorsque le traitement n’est pas associé à une thérapie complémentaire. Ceci est aussi valable pour d’autres substances comme par exemple la ciclosporine A. L’ancien antidépresseur, doxépine (Sinquan ®) est une substance intéressante dans la lutte contre le prurit. Ce médicament a un effet systémique sur les mécanismes centraux des différentes formes de démangeaisons, mais il a probablement aussi des effets sur les nerfs cutanés, puisqu’un traitement topique peut également réduire le prurit.
La doxépine topique peut être appliquée seule ou en combinaison avec la capsaïcine.14 La capsaïcine, tirée de gousses de poivrier, a une influence directe sur les terminaisons nerveuses et vide tous les réservoirs de substances P dans la peau. Les douleurs, avec sensations de brûlures, limitent l’application de la capsaïcine. Mis à part la capsaïcine, le polidocanol est encore utilisé de façon topique, mais son efficacité antiprurigineuse n’aurait été prouvée dans aucune étude contrôlée. Les stéroïdes topiques ont un effet anti-inflammatoire et par conséquent peuvent être utilisés uniquement dans ce cas-là. L’effet antiprurigineux des stéroïdes locaux est dans la plupart des cas très court et en raison des effets secondaires, ces médicaments devraient être administrés de façon ciblée et limitée lors du traitement des démangeaisons. L’application des nouveaux inhibiteurs de la calcineurine (Tacrolimus ®, Pimecrolimus ®) pourrait être intéressante.15 Des indications révèlent que ces médicaments développent non seulement des effets antiprurigineux avec diminution de l’inflammation, mais peuvent également influencer la fonction cutanée des terminaisons nerveuses.16 Pratiquement, toutes les applications topiques contre le prurit ont des désavantages, leur efficacité n’a pas encore été prouvée par des études précises double aveugle et placebo contrôlé. Dans les études ouvertes, l’effet antiprurigineux des bases hydratantes grasses ne pourra jamais être exclu avec certitude.
Le principal problème de la dermatite atopique c’est le prurit chronique et aigu combiné avec une peau sèche et craquelée (figure 2). La thérapie n’est pas simple et avec des mesures anti-inflammatoires uniques, seule la partie enflammée peut être traitée et l’effet antiprurigineux est dans la plupart des cas de courte durée. Le tableau 4 montre un schéma thérapeutique possible lors de dermatite atopique avec des démangeaisons chroniques. Une des premières mesures, mais aussi la plus importante, est la thérapie de soins hydratants de crème grasse. Selon le degré d’importance de la symptomatologie, on pourra combiner le traitement avec des antihistaminiques (antihistaminiques non sédatifs le matin, antihistaminiques sédatifs le soir). On peut prendre à court terme des stéroïdes topiques ou des inhibiteurs de calcineurine. Au cas où ces mesures ne devaient pas suffire, on pourrait y ajouter une UV-thérapie (arrêter les inhibiteurs de calcineurine). Par la suite, on peut envisager l’utilisation orale de doxépine ou de néméxine. Lors de démangeaisons chroniques, il est important de maintenir la thérapie antiprurigineuse durant plusieurs mois afin d’éviter une récidive.


Chaque thérapie antiprurigineuse est une thérapie sur mesure et doit être adaptée à chaque patient. La complexité de cette thérapie, ainsi que le manque de stratégies renouvelées et innovatrices montrent qu’il y a encore beaucoup à faire dans la recherche fondamentale et clinique dans le domaine du prurit. Les entreprises sont également sollicitées à investir dans la recherche de nouveaux concepts thérapeutiques. Des indications précises et un mode d’application pour de nouvelles substances potentiellement efficaces, telles que les cannabinoïdes, les anesthésiants locaux ou les antagonistes du récepteur opioïde doivent être examinés dans le cadre d’études cliniques. Au Département de dermatologie de l’Hôpital universitaire de Lausanne, nous essayons de combiner nos expériences de recherche fondamentale, de recherche et d’expérience clinique. Notre laboratoire de recherche se consacre déjà depuis plusieurs années à la neurodermatologie. Notre principal intérêt est de connaître l’influence du système récepteur-opioïde sur le système nerveux cutané et le prurit.3,17-19 Sur la base de résultats de la recherche fondamentale, nous achevons avec succès une première étude clinique. Afin de pouvoir rassembler de précieuses expériences dans le traitement de la résistance des différentes formes de démangeaisons, nous avons décidé de créer une consultation des démangeaisons, analogue à la consultation de la douleur. Ces efforts, de tendance internationale, ont pour but de considérer le problème des démangeaisons comme sérieux et de trouver des solutions conformes au bien-être des patients. |
Auteur(s) : P. L. Bigliardi
Contact de(s) l’auteur(s) : Dr Paul L. Bigliardi Service de dermatologie CHUV, 1011 Lausanne Paul.bigliardi@chuv.ch
Bibliographie : 1 ** Oaklander AL, Siegel SM. Cutaneous innervation : Form and function. J Am Acad Dermatol 2005;53: 1027-37. 2 * Zylka MJ, Rice FL, Anderson DJ. Topographically distinct epidermal nociceptive circuits revealed by axonal tracers targeted to Mrgprd. Neuron 2005;45:17-25. 3 ** Bigliardi-Qi M, Lipp B, Sumanovski LT, et al. Changes of epidermal mu-opiate receptor expression and nerve endings in chronic atopic dermatitis. Dermatology 2005;210:91-9. 4 * Schmelz M, Schmidt R, Bickel A, et al. Specific C-receptors for itch in human skin. J Neurosci 1997;17: 8003-8. 5 * Stander S, Steinhoff M, Schmelz M, et al. Neurophysiology of pruritus : Cutaneous elicitation of itch. Arch Dermatol 2003;139:1463-70. 6 Orstavik K, Weidner C, Schmidt R, et al. Pathological C-fibres in patients with a chronic painful condition. Brain 2003;126:567-78. 7 Heyer GR, Hornstein OP. Recent studies of cutaneous nociception in atopic and non-atopic subjects. J Dermatol 1999;26:77-86. 8 * Ward JR, Bernhard JD. Willan’s itch and other causes of pruritus in the elderly. Int J Dermatol 2005; 44:267-73. 9 ** Wahlgren CF. Itch and atopic dermatitis : An overview. J Dermatol 1999;26:770-9. 10 * Szepietowski JC, Sikora M, Kusztal M, et al. Uremic pruritus : A clinical study of maintenance hemodialysis patients. J Dermatol 2002;29:621-7. 11 Bergasa NV, Thomas DA, Vergalla J, et al. Plasma from patients with the pruritus of cholestasis induces opioid receptor-mediated scratching in monkeys. Life Sci 1993;53:1253-7. 12 Bircher AJ, Meier-Ruge W. Aquagenic pruritus. Water-induced activation of acetylcholinesterase. Arch Dermatol 1988;124:84-9. 13 Stander S, Szepfalusi Z, Bohle B, et al. Differential storage of hydroxyethyl starch (HES) in the skin : An immunoelectron-microscopical long-term study. Cell Tissue Res 2001;304:261-9. 14 McCleane G. Topical application of doxepin hydrochloride, capsaicin and a combination of both produces analgesia in chronic human neuropathic pain : A randomized, double-blind, placebo-controlled study. Br J Clin Pharmacol 2000;49:574-9. 15 Aguilar-Bernier M, Bassas-Vila J, Sanz-Munoz C, et al. Successful treatment of pruritus with topical tacrolimus in a patient with primary biliary cirrhosis. Br J Dermatol 2005;152:808-9. 16 Stander S, Luger TA. (Antipruritic effects of pimecrolimus and tacrolimus). Hautarzt 2003;54:413-7. 17 Bigliardi PL, Bigliardi-Qi M, Peripheral Opiate Receptor System in Human Epidermis and Itch, in Itch : Basic Mechanisms and Therapy, G. Yosipovitch, et al., Editors. Basel : Marcel Dekker, Inc. : New York, 2004;97-106. 18 Bigliardi-Qi M, Sumanovski LT, Buchner S, et al. Mu-opiate receptor and Beta-endorphin expression in nerve endings and keratinocytes in human skin. Dermatology 2004;209:183-9. 19 ** Biro T, Ko MC, Bromm B, et al. How best to fight that nasty itch – from new insights into the neuroimmunological, neuroendocrine, and neurophysiological bases of pruritus to novel therapeutic approaches. Exp Dermatol 2005;14:225-40. * à lire ** à lire absolument
Mots-clef :
Numéro de revue : 63
Numéro d’article : 31231 |












































{kind=link}